The complete human genome has been gradually revealed.
Translator: @Anh-Vu Mai-Nguyen
Advisor: @Thanh Nguyen @Nam Sy Vo
On May 27, 2021, researchers from the Telomere-to-Telomere Consortium decoded an additional 200 million bases and 115 protein-coding regions across most chromosomes compared to the current reference genome. This is considered the next major achievement in human genome sequencing since 2013.
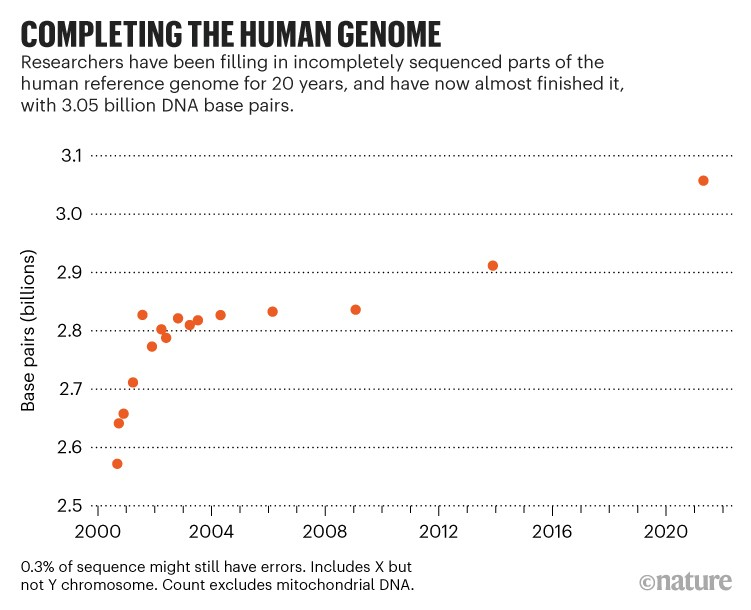
In the 20 years since the first human genome sequencing project was launched by the Human Genome Project and the biotechnology company Celera Genomics, the human genome has continued to be completed and updated. At that time, it was estimated that about 15% of the entire genome had not been successfully sequenced due to technological limitations, preventing researchers from finding the exact location of some DNA segments, especially repeat gene regions. Thanks to the relentless efforts, scientists have made some significant progress: reducing the proportion of missing regions to 8% of the GRCh38 reference genome – released in 2013.
Recently, researchers in the Telomere-to-Telomere Consortium (T2T Consortium) – an international collaboration of about 30 different organizations, have identified a large number of missing regions. In a preprint published on May 27, 2021, titled “The complete sequence of a human genome,” geneticist Karen Miga and colleagues at the University of California – Santa Cruz said they had successfully decoded the remaining 8%, thereby discovering about 115 new genes, out of a total of 19,969 genes.
“It’s great to have a solution for complex regions,” said Kim Pruitt, a bioinformatician at the National Center for Biotechnology Information (NCBI) in Bethesda, Maryland. She called the results “a remarkable milestone.”
New sequencing technology
The newly sequenced genome – called T2T-CHM13 – adds nearly 200 million base pairs to the 2013 version of the human genome.
Unlike previous attempts, instead of taking DNA samples directly from humans, the researchers used a cell line derived from thai trứng (chửa trứng), a type of tissue that forms in humans when a sperm fertilizes an egg without a nucleus. The resulting cell contains only the chromosomes from the father, so researchers do not need to distinguish between two sets of chromosomes from two different people (father and mother).
Miga believes that new sequencing technology (link to sequencing technology article) from Pacific Biosciences in Menlo Park, California, played a particularly important role in this discovery. PacBio’s technology uses a laser to scan long stretches of DNA (up to 20,000 bases) extracted from cells. Traditional technologies usually read only short stretches of DNA of a few hundred bases, and then assemble these fragments like puzzle pieces. Assembling long fragments is often much simpler, because they contain many overlapping segments – a feature that makes assembly easier.
However, T2T-CHM13 is not the final reference genome. The T2T team still had trouble resolving some regions, and estimated that about 0.3 percent of the genome may still contain errors. Although there are no more blank spaces in the genome, quality assurance on these error regions is still relatively difficult, said author Miga. In addition, the sperm cells used to form the fertilized egg carry the X chromosome, so researchers have not been able to sequence the Y chromosome – which normally triggers biological development in males.
Hundreds of other genomes are ready to be sequenced.
Even complete, T2T-CHM13 represents only one person’s genome. The T2T Consortium has partnered with the Human Pangenome Reference Consortium, which aims to sequence more than 300 human genomes from around the world over the next three years. Miga says other groups could use T2T-CHM13 to reference parts of the genome that differ between individuals. Miga’s team also plans to sequence entire genomes containing both maternal and paternal chromosomes, and is currently sequencing the Y chromosome, using the same new methods to find missing regions. Miga expects that geneticists will quickly determine whether these newly sequenced regions are linked to human diseases. “When the human genome first came out, we didn’t have the tools, but now we can get information about the function of newly sequenced genes much faster, because we have so many resources at our disposal,” she says.
Miga hopes that future human genome sequences will include all the newly sequenced parts—not just the easy-to-read parts. This will become easier once the reference genome is complete and the technical hurdles are ironed out. “We need to move toward a new standard in genomics, where these techniques become routine rather than something special,” she says.
Vietnam is also building its own reference genome.
Although the complete genome of T2T-CHM13 has given us a more comprehensive view of the human genome, it cannot yet replace GRCh38 due to the lack of accompanying annotation data sets. Bioinformatics research therefore has to continue to cope with the limitations of GRCh38, such as the genome being too biased towards Europeans and Americans, and the lack of short p arms of autosomal chromosomes (13, 15, 21, 22), etc. In recent years, especially Asian countries such as Korea and Japan have invested in and built their own reference genomes, taking a step further towards precision medicine. Japan has updated its reference genome twice, and with the most recent publication in January 2021, the Japanese reference genome has reached a size of 3.08 billion bases, with only nearly 500 gaps left. To achieve this, the research team in Japan used a combination of DNA from 3 volunteers and 5 different sequencing technologies.
Currently, VinBigData has planned to build and annotate a reference genome of the Vietnamese people (link to the Vietnamese Reference Genome project). This project will utilize data from projects that have been implemented at VinBigdata (link to Data Portal) and the most advanced sequencing technologies to create the first nearly complete reference genome of the Vietnamese people. The project also has cooperation with leading domestic and international research units such as Hanoi Medical University, University of Queensland, Australia, University of California, San Diego, USA. Following the success of regional and international reference genome studies, this project promises to provide a useful reference source for genetic studies on the Vietnamese population.
References:
A complete human genome sequence is close: how scientists filled in the gaps